
Le Degenerazioni tapeto-retiniche comprendono un ampio capitolo della patologia retinica e poichè ancora molte sono le incertezze sussistenti circa alcuni aspetti patogenetici, la classificazione schematica più agevole può essere proposta in base al livello lesionale primario.
Sono da distinguere pertanto
- le lesioni che interessano primitivamente l'epitelio pigmentato
- quelle che interessano l'epitelio pigmentato e neuroepitelio
- le lesioni che coinvolgono lo strato nervoso retinico.
Questa suddivisione utile da un punto di vista diagnostico-terapeutico è poco valida dal punto di vista patologico-evolutivo della malattia data la stretta correlazione esistente tra i vari foglietti e pertanto la lesione dell'uno si potrà far risentire, seppure con espressività diversa secondo la qualità della lesione primaria sui restanti.
Le degenerazioni dell'epitelio pigmentato possono essere sia ereditarie che acquisite.
Tra le forme ereditarie si possono avere lesioni diffuse e lesioni localizzate alla retina centrale.
In entrambi i casi l'alterazione consiste in un deposito di mucopolisaccaridi acidi nella cavità interna delle cellule dell'epitelio pigmentato che sostituiscono i granuli pigmentati citoplasmatici.
Le forme acquisite sono dovute all'infezione virale rubeolica.
Il virus rubeolico può esplicare un'azione lesiva sull'epitelio pigmentato durante le prime settimane di vita intrauterina. Tale virus provoca la necrosi di gruppi cellulari, pertanto queste cellule risultano sovraccariche di melanina.
Tra le Degenerazioni tapeto-retiniche la più diffusa è la Retinite Pigmentosa:

patologia degenerativa bilaterale, geneticamente determinata ed a lento decorso.
Le alterazioni iniziali sono a carico dell'epitelio pigmentato e degli articoli esterni dei bastoncelli, in un secondo momento vengono colpiti anche i coni; i rimanenti strati nervosi sono compromessi solo tardivamente.
Le lesioni interessano per prima la periferia retroequatoriale per poi generalizzarsi a tutto l'ambito retinico.
L'epitelio pigmentato subisce modificazioni di forma, consistenti in appiattimento e metaplasia fusiforme con depigmentazione.
Anche la membrana di Bruch presenta lesioni involutive quale conseguenza di scarichi metabolici anormali derivanti dagli strati retinici alterati. Successivamente le cellule dell'epitelio pigmentato migrano verso gli strati più interni della retina, raggruppandosi in ammassi che presentano al centro una sostanza ialina prodotta dalle cellule stesse.
Gli articoli esterni dei bastoncelli sono i primi ad essere compromessi, si raccorciano e dissolvendosi sono rimpiazzati da materiale eosinofilo. I vasi retinici subiscono un ispessimento della parete ed una degenerazione ialina. I vasi coroideali invece vengono risparmiati.
Varianti della Retinite Pigmentosa sono:
- la Malattia di Leber
- la Retinite Puntata Albescente
- la Malattia di Oguchi.
Il diverso grado di mobilizzazione delle cellule pigmentate negli strati nervosi retinici è la caratteristica che differenzia queste forme dalla Degenerazione tapeto-retinica classica.
Malattia di Leber:
la degenerazione neuro epiteliale è più precoce rispetto a quella dell'epitelio pigmentato, inizia infatti in fase prenatale. Questa precoce lesione funzionale coinvolge anche la porzione retinica centrale.
Retinite Puntata Albescente:
in questa forma alla dispersione pigmentaria si sostituiscono piccoli puntini bianchi che non coinvolgono però la regione maculare. Clinicamente si distinguono due forme: una progressiva l'altra stazionaria. Una variante particolare della forma stazionaria è la Malattia di Oguchi. In tale forma la retina ha la tipica colorazione rossastra solo dopo un periodo di oscuramento, mentre si presenta decolorata dopo l'abbagliamento.
Malattia di Refsum:
tipica per la sua alterazione congenita del metabolismo dei lipidi, con accumulo di acido fittanico nel sangue e nelle cellule, in particolare nelle cellule dell'epitelio pigmentato. Clinicamente si associa a sordità centrale, polinevrite, atassia.
Il sintomo più evidente è l'emeralopia, ovvero la difficoltà ingravescente che il paziente mostra nella visione scotopica. L'emeralopia è caratterizzata da un innalzamento della soglia luminosa e da una rallentata capacità di adattamento al buio. Questo disturbo compare abitualmente nell'infanzia generalmente i genitori si accorgono che questi bambini quando passano da un ambiente illuminato ad un ambiente buio o scarsamente illuminato, presentano un'enorme difficoltà ad orientarsi. A tale disturbo si affianca in un secondo momento una discromatopsia, dapprima parziale per il verde e poi totale.
Le alterazioni elettrofisiologiche assumono un'importanza notevole; compromesse risultano le risposte elettroretinografiche nelle componenti positive, rappresentate fondamentalmente da un marcato ipovoltaggio dell'ERG che nel proseguo evolutivo non sono più registrabili, giungendo all'estinzione. Di fondamentale considerazione è l'abbassamento dell'indice di Arden e la curva piatta che fornisce l'EOG.
Non meno considerate devono essere le alterazioni del campo visivo, rappresentate da un deficit anulare, che partendo dalla macchia cieca, abbraccia l'area centrale del campo visivo separandola dai settori periferici.
Lo scotoma, col proseguire degli anni si allarga sempre più, estendendosi soprattutto nelle regioni periferiche tanto che da uno stadio più avanzato, il campo visivo è ridotto ad un isolotto centrale compreso tra le isoptere di 10° e 15°, definito "tubulare".
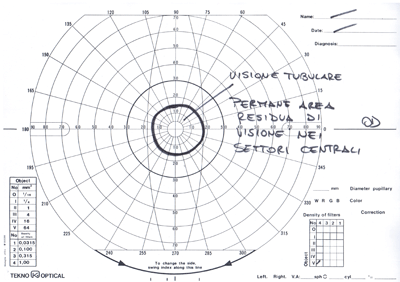
In queste condizioni, anche se l'acuità visiva è ancora buona, il paziente trova difficoltà nell'orientarsi, per la limitazione del campo visivo che lo pone nella condizione di chi guardasse attraverso le canne di un fucile.
Successivamente il coinvolgimento maculare può danneggiare irreparabilmente anche la visione centrale.
Nelle forme tardive l'evoluzione è benigna e una discreta funzione visiva può permanere per tutta la vita.
Le recenti acquisizioni presenti in letteratura sottolineano l'importanza che bisogna dare in special modo all'elettrofisiologia e alla campimetria in quanto possono anticipare la comparsa delle alterazioni del fondo oculare in considerazione del fatto che in un primo momento il fundus di entrambi gli occhi può apparire normale e solo in un secondo momento molti pazienti presentano il tipico disturbo della rarefazione e accumulo di pigmento a sale e pepe.
Le arteriole retiniche si assottigliano e talvolta per obliterazione del lume vasale, assumono l'aspetto di cordoni biancastri e la papilla diventa pallida sino a raggiungere un colorito cereo.
Oftalmoscopicamente si potranno osservare numerose chiazze nerastre a bordi regolari e ramificati: accumuli di pigmento osteocitiformi.
La correlazione tra le alterazioni elettrofisiologiche e perimetriche, riveste una particolare importanza prognostica sull'aggressività della patologia, anche in relazione all'età di insorgenza della medesima.
I soggetti gravemente compromessi nella capacità visiva come pure nell'orizzonte percepito, per le amputazioni scotomatose per le degenerazioni da autolisi, possono necessitare di ausili per ipovisione.
Molto spesso i pazienti, pur bisognosi di quest'approccio non lo accettano per un handicap anche psicologico.
Riveste così un'importanza fondamentale la collaborazione con uno psicologo per un sostegno mirato ad un'integrazione non solo delle residue capacità visive, ma con tutto l'"io" e la personalità del soggetto affetto da un così grave handicap.